1-4 分析框架和工具¶
我们在前面的分析中已经获得了样本的计数矩阵(cells x genes),我们需要从计数矩阵中,挖掘潜在的细胞或者基因信息。由于数据规模往往较大,常规的Pandas,Numpy无法同时容纳不同维度的信息,这对分析工具提出了新的要求。目前单细胞领域的工具主要有三种:
- Bioconductor:R语言实现的生物信息学生态
- Seurat:R语言实现的单细胞分析生态
- Scverse:基于Python实现的单细胞分析生态。
Bioconductor 是一个开发、支持和共享免费开源软件的项目,重点是对包括单细胞在内的许多不同生物测定的数据进行严格且可重复的分析。同质的开发人员和用户体验以及带有用户友好小插图的丰富文档是 Bioconductor 的最大优势。Seurat 是一款备受推崇的 R 软件包,专为分析单细胞数据而设计。它为分析的所有步骤提供工具,包括多模式和空间数据。修拉以写得好的小插图和庞大的用户群而闻名。然而,对于极大的数据集(超过 50 万个单元),这两种 R 选项都会遇到可扩展性问题,这促使基于 Python 的社区开发 scverse 生态系统。scverse 是一个致力于生命科学基础工具的组织和生态系统,最初重点关注单细胞。可扩展性、可扩展性以及与现有 Python 数据和机器学习工具的强大互操作性是 scverse 生态系统的一些优势。
而在我们的教程中,我们还会介绍omicverse框架,该框架整合了大量RNA-seq的处理算法,并重新定义了不同算法的数据格式,提升了兼容性,更具有用户友好的特征。此外,omicverse还能完成CNS级别的可视化图表的绘制。基于omicverse框架我们可以完成更多单细胞领域探索的任务。
在本章中,我们将介绍scverse生态中所包含的数据结构,包括AnnData,MuData。二者是我们使用Python进行单细胞分析所涉及的基本数据结构。
1. 使用AnnData存储数据¶
我们在此前的学习中,掌握了Pandas的数据格式与操作。而AnnData,则是基于Pandas的基本格式,赋予了更多层的数据含义。在Pandas中,我们的数据由index与columns进行索引,但是对于index与columns自身所包含的信息,则需要额外的文件进行存储和补充。例如,我们的计数矩阵的格式为(cells x genes),我们可以定义columns为cells,index为genes,对于cells,我们会关注每一个细胞是什么类型的细胞,每一个细胞里表达的基因数量;对于genes,我们会关注每一个基因的id,基因名,染色体位置(chr:10000-20000)等。故我们需要一种包含更多维度信息的数据格式。
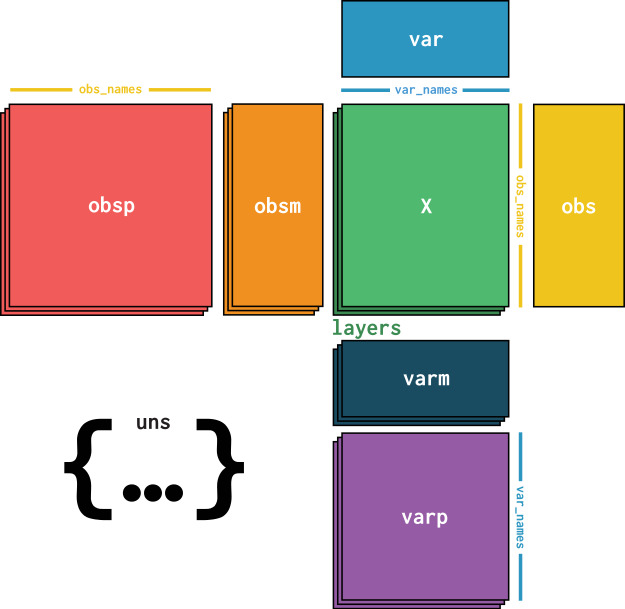
在这里,我们介绍AnnData格式,该数据的格式如图所示,以计数矩阵为核心,向外扩展出了obs,var两个数据层,并且obs和var还可以存放更进一步的数据内容。同时计数矩阵由layers叠加,我们可以存放不同类型的计数矩阵,如归一化后的计数矩阵,原始计数矩阵等。我们还可以在uns层中存放其他任何你想存放的数据。
1.1 安装¶
AnnData 可在 PyPI 或 Conda上使用,并且可以使用以下任一方法安装:
%pip install anndata
%conda install -c conda-forge anndata
1.2 初始化AnnData对象¶
本节的灵感来自 AnnData 的“入门”教程:https://anndata-tutorials.readthedocs.io/en/latest/getting-started.html
让我们创建一个具有稀疏计数矩阵的简单 AnnData 对象,例如可以表示基因表达计数。首先,我们导入所需的包。
import numpy as np
import pandas as pd
import anndata as ad
from scipy.sparse import csr_matrix
下一步,我们使用随机泊松分布数据初始化 AnnData 对象。一般来说,我们会把AnnData实例化的对象叫做adata
counts = csr_matrix(np.random.poisson(1, size=(100, 2000)), dtype=np.float32)
adata = ad.AnnData(counts)
adata
AnnData object with n_obs × n_vars = 100 × 2000
获得的AnnData对象有100个obs和2000个var。这相当于 100 个细胞和 2000 个基因。我们可以使用adata.X来访问我们的计数矩阵。
adata.X
<100x2000 sparse matrix of type '<class 'numpy.float32'>' with 126647 stored elements in Compressed Sparse Row format>
我们可以使用.obs_names和.var_names为obs和var提供索引,当然你也可以使用obs.index,var.index,二者的效果是一样的。
adata.obs_names = [f"Cell_{i:d}" for i in range(adata.n_obs)]
adata.var_names = [f"Gene_{i:d}" for i in range(adata.n_vars)]
print(adata.obs_names[:10])
Index(['Cell_0', 'Cell_1', 'Cell_2', 'Cell_3', 'Cell_4', 'Cell_5', 'Cell_6',
'Cell_7', 'Cell_8', 'Cell_9'],
dtype='object')
#随机生成细胞类型,并存放到变量ct中
ct = np.random.choice(["B", "T", "Monocyte"], size=(adata.n_obs,))
#我们将生成的细胞类型赋值到adata.obs列中
adata.obs["cell_type"] = pd.Categorical(ct) # Categoricals are preferred for efficiency
adata.obs
| cell_type | |
|---|---|
| Cell_0 | B |
| Cell_1 | Monocyte |
| Cell_2 | Monocyte |
| Cell_3 | T |
| Cell_4 | T |
| ... | ... |
| Cell_95 | Monocyte |
| Cell_96 | B |
| Cell_97 | B |
| Cell_98 | B |
| Cell_99 | Monocyte |
100 rows × 1 columns
如果我们现在再次检查 AnnData 对象的内容,我们将注意到它也被更新了,在 obs 中包含 cell_type 信息。
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
bdata = adata[adata.obs.cell_type == "B"]
bdata
View of AnnData object with n_obs × n_vars = 32 × 2000
obs: 'cell_type'
1.4 Observation/variable 的矩阵¶
我们注意到,在.obs或.var中,我们可以存放细胞类型的元数据,但是这个元数据是一维的,如果我们有一个特征是多维的呢?比如细胞的特征向量,这时候,我们可以将数据存放在.obsm或者.varm中,表示多维的元数据。
让我们从一个随机生成的矩阵开始,我们可以将其解释为细胞的特征向量UMAP,我们还可以随机生成一个矩阵表示基因的一些多维元数据特征。
adata.obsm["X_umap"] = np.random.normal(0, 1, size=(adata.n_obs, 2))
adata.varm["gene_stuff"] = np.random.normal(0, 1, size=(adata.n_vars, 5))
adata.obsm
AxisArrays with keys: X_umap
同样的,AnnData对象也被更新了
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
obsm: 'X_umap'
varm: 'gene_stuff'
这里有一些关于.obsm/.varm的额外信息:
- 多维的元数据可以是
pandas的DataFrames格式,也可以是numpy的ndarray,还可以是scipy的sparse matrix - 元数据可以很方便地被
scanpy识别,进行绘图操作
1.5 非结构化元数据¶
如上所述,AnnData 有.uns,它允许任何非结构化元数据。这可以是任何东西,例如包含一些对数据分析有用的一般信息的列表或字典。尝试仅将此插槽用于无法有效存储在其他插槽中的数据。
adata.uns["random"] = [1, 2, 3]
adata.uns
OverloadedDict, wrapping:
OrderedDict([('random', [1, 2, 3])])
With overloaded keys:
['neighbors'].
1.6 Layers¶
最后,我们可能有不同形式的原始核心数据,可能一种是标准化的,另一种不是标准化的。这些可以存储在 AnnData 的不同层中。
例如,让我们对原始数据进行对数转换并将其存储在layers中。
adata.layers["log_transformed"] = np.log1p(adata.X)
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
uns: 'random'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'
我们的原始矩阵X没有修改并且仍然可以访问。我们可以通过比较原始layers:X和新layers:log_transformed来验证这一点。
(adata.X != adata.layers["log_transformed"]).nnz == 0
False
1.7 导出为DataFrames¶
我们有时候需要导出pandas的DataFrames来上传到其他工具进行分析,也就是计数矩阵,我们可以很轻松地实现这点,在AnnData中。我们可以导出任意layers,如果不指定则导出.X
adata.to_df(layer="log_transformed").head()
| Gene_0 | Gene_1 | Gene_2 | Gene_3 | Gene_4 | Gene_5 | Gene_6 | Gene_7 | Gene_8 | Gene_9 | ... | Gene_1990 | Gene_1991 | Gene_1992 | Gene_1993 | Gene_1994 | Gene_1995 | Gene_1996 | Gene_1997 | Gene_1998 | Gene_1999 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell_0 | 0.000000 | 1.386294 | 0.693147 | 0.693147 | 0.693147 | 0.000000 | 0.693147 | 0.000000 | 0.693147 | 1.098612 | ... | 0.000000 | 1.386294 | 0.693147 | 0.000000 | 0.693147 | 1.386294 | 0.693147 | 1.098612 | 0.693147 | 0.000000 |
| Cell_1 | 0.000000 | 0.000000 | 1.609438 | 0.693147 | 0.000000 | 0.693147 | 0.693147 | 0.000000 | 1.098612 | 1.098612 | ... | 0.000000 | 0.693147 | 0.693147 | 1.609438 | 0.000000 | 0.000000 | 0.693147 | 0.000000 | 0.693147 | 0.693147 |
| Cell_2 | 1.098612 | 0.693147 | 0.000000 | 0.693147 | 0.693147 | 1.386294 | 1.098612 | 0.693147 | 0.000000 | 1.098612 | ... | 1.609438 | 0.000000 | 1.098612 | 0.693147 | 0.000000 | 0.000000 | 1.098612 | 1.098612 | 0.693147 | 0.000000 |
| Cell_3 | 0.693147 | 0.000000 | 0.000000 | 0.693147 | 0.693147 | 0.000000 | 0.693147 | 0.693147 | 0.693147 | 0.693147 | ... | 0.693147 | 0.693147 | 0.693147 | 0.693147 | 0.693147 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.693147 |
| Cell_4 | 0.693147 | 0.000000 | 1.386294 | 0.693147 | 1.098612 | 1.098612 | 0.000000 | 0.000000 | 0.693147 | 0.693147 | ... | 1.386294 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 0.000000 | 0.693147 | 0.000000 | 0.693147 | 0.000000 |
5 rows × 2000 columns
1.8 AnnData 的读写¶
AnnData 对象可以保存在磁盘上的分层数组存储(如HDF5或Zarr)中,以在磁盘和内存中启用类似的结构。AnnData 带有自己的基于 HDF5 的持久文件格式:h5ad. 如果类别数量较少的字符串列尚未分类,AnnData 会自动将它们转换为分类。现在,我们将以格式保存 AnnData 对象h5ad。
adata.write("../../data/my_results.h5ad", compression="gzip")
!h5ls '../../data/my_results.h5ad'
X Group layers Group obs Group obsm Group obsp Group uns Group var Group varm Group varp Group
……然后读回来。
adata_new = ad.read_h5ad("../../data/my_results.h5ad")
adata_new
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
uns: 'random'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'
obs_meta = pd.DataFrame(
{
"time_yr": np.random.choice([0, 2, 4, 8], adata.n_obs),
"subject_id": np.random.choice(
["subject 1", "subject 2", "subject 4", "subject 8"], adata.n_obs
),
"instrument_type": np.random.choice(["type a", "type b"], adata.n_obs),
"site": np.random.choice(["site x", "site y"], adata.n_obs),
},
index=adata.obs.index, # these are the same IDs of observations as above!
)
我们使用该obs_meta取代原来AnnData对象的.obs,为了避免混淆,我们生成一个新的AnnData对象
adata = ad.AnnData(adata.X, obs=obs_meta, var=adata.var)
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
需要注意的是,与numpy类似,我们所浏览的是AnnData的视图,并不会直接显示变量,这样可以避免额外的内存开销。
此外,当我们对AnnData进行任何修改时,都会在函数内部调用.copy()完成AnnData实际内容的修改。我们也可以直接使用.copy()来获取AnnData的实际变量,但通常该操作的意义不大,除了在某些特定场景的函数中,比如scvi-tools中的模型训练中,会用到AnnData的实际内容。
我们使用.[]来对AnnData对象进行切片操作,与pandas不同的是,我们在.[]中输入整数则类似.iloc,输入字符串或者逻辑值则类似.loc,例如,我想查看前五个细胞,以及基因1和基因3的AnnData对象的子集。
adata_view = adata[:5, ["Gene_1", "Gene_3"]]
adata_view
View of AnnData object with n_obs × n_vars = 5 × 2
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
我们发现View of AnnData object with n_obs × n_vars = 5 × 2,但实际上的AnnData并没有被修改,这与pandas是一致的
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
如果我们想要一个 AnnData 将数据保存在内存中,则必须调用.copy().
adata_subset = adata[:5, ["Gene_1", "Gene_3"]].copy()
adata_subset
AnnData object with n_obs × n_vars = 5 × 2
obs: 'time_yr', 'subject_id', 'instrument_type', 'site'
为什么强调这个概念,是因为当你修改adata_subset的时候,原adata不会被修改,但当你修改adata_view的时候,原adata的相应内容也会被修改,这可能会引发一些逻辑错误。
对于View,我们还可以设置列的前 3 个元素。我们将前3个细胞的基因1的表达量设置成0
print(adata[:3, "Gene_1"].X.toarray().tolist())
adata[:3, "Gene_1"].X = [0, 0, 0]
print(adata[:3, "Gene_1"].X.toarray().tolist())
[[3.0], [0.0], [1.0]] [[0.0], [0.0], [0.0]]
虽然前面强调了adata_view和adata_subset的不同,但实际上,我们如果修改adata_view的内容的时候,会自动调用.copy使数据存放到内存上
adata_view.obs["foo"] = range(5)
/var/folders/4m/2xw3_2s503s9r616083n7w440000gn/T/ipykernel_2195/3248193034.py:1: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual. adata_view.obs["foo"] = range(5)
现在adata_view存储实际数据,不再只是对 adata 的引用。
adata_view
AnnData object with n_obs × n_vars = 5 × 2
obs: 'time_yr', 'subject_id', 'instrument_type', 'site', 'foo'
当然,我们也可以使用pandas的bool索引来进行AnnData的切片操作
adata[adata.obs.time_yr.isin([2, 4])].obs.head()
| time_yr | subject_id | instrument_type | site | |
|---|---|---|---|---|
| Cell_0 | 2 | subject 8 | type a | site x |
| Cell_7 | 4 | subject 1 | type b | site y |
| Cell_8 | 4 | subject 2 | type a | site x |
| Cell_11 | 4 | subject 1 | type a | site x |
| Cell_16 | 2 | subject 1 | type b | site x |
adata = ad.read("../../data/my_results.h5ad", backed="r")
adata.isbacked
True
如果这样做,则需要记住 AnnData 对象与用于读取的文件有一个连接是为关闭的,这类似于我们打开一个文件的意思。
adata.filename
PosixPath('../../data/my_results.h5ad')
因为我们在只读模式下使用它,所以我们不能损坏任何东西。要继续学习本教程,我们仍然需要显式地关闭它。
adata.file.close()
2. 使用MuData存放多模态的数据¶
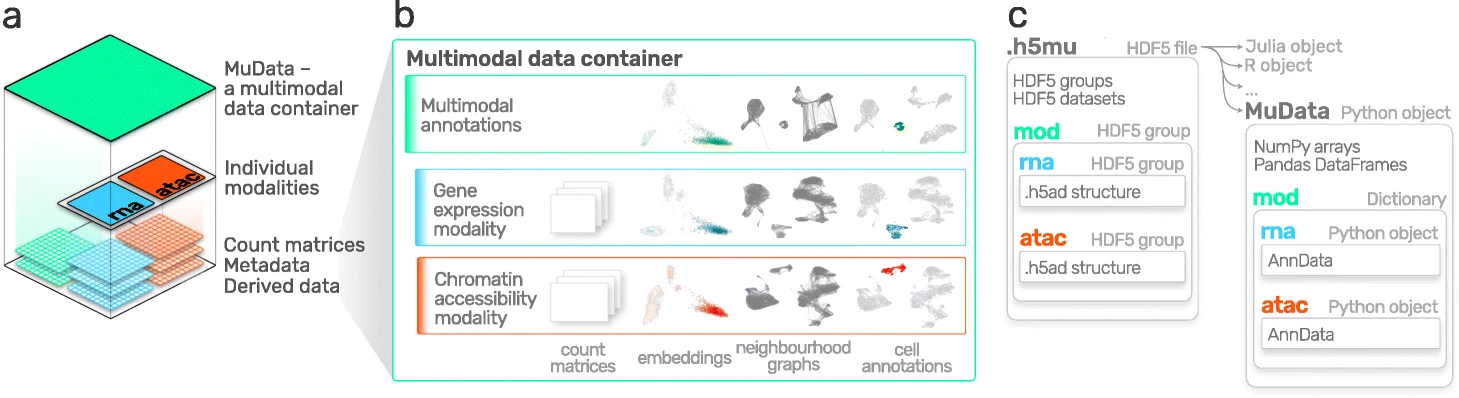
AnnData 主要用于存储和操作单模态数据。然而,CITE-Seq 等单细胞多模态计数通过同时测量 RNA 和表面蛋白来生成多模态数据。
这些数据需要更先进的存储方式,这就是 MuData 发挥作用的地方。MuData 构建在 AnnData 之上,用于存储和操作多模态数据。
2.1 安装¶
MuData 可在 PyPI 或 Conda上使用,并且可以使用以下任一方法安装:
%pip install mudata
%conda install -c conda-forge mudata
MuData 背后的主要思想:MuData 对象包含对单模态数据的单个 AnnData 对象的引用,但 MuData 对象本身也存储多模态注释。因此,可以直接访问 AnnData 对象来执行单模态数据转换,将其结果存储在相应的 AnnData 注释中,而且还可以聚合联合计算的模态,其结果可以存储在全局 MuData 对象中。
从技术上讲,这是通过 MuData 对象实现的,该对象包含一个带有 AnnData 对象的字典,每个模态一个,在其.mod(=modality) 属性中。正如 AnnData 对象本身一样,它们也包含诸如.obs或者var(样本或细胞)之类的属性,.obsm及.varm其多维注释,例如嵌入。
2.2 初始化MuData对象¶
我们将从 mudata 包中导入 MuData 开始。
import mudata as md
#为了创建MuData,我们需要模拟生成一些数据
n, d, k = 1000, 100, 10
z = np.random.normal(loc=np.arange(k), scale=np.arange(k) * 2, size=(n, k))
w = np.random.normal(size=(d, k))
y = np.dot(z, w.T)
y.shape
(1000, 100)
要创建 MuData 对象,我们首先需要多个单模态 AnnData 对象。因此,我们创建两个 AnnData 对象,其中包含相同obs值但不同var的数据。
adata = ad.AnnData(y)
adata.obs_names = [f"obs_{i+1}" for i in range(n)]
adata.var_names = [f"var_{j+1}" for j in range(d)]
adata
/var/folders/4m/2xw3_2s503s9r616083n7w440000gn/T/ipykernel_2195/3341965559.py:1: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour. adata = ad.AnnData(y)
AnnData object with n_obs × n_vars = 1000 × 100
d2 = 50
w2 = np.random.normal(size=(d2, k))
y2 = np.dot(z, w2.T)
adata2 = ad.AnnData(y2)
adata2.obs_names = [f"obs_{i+1}" for i in range(n)]
adata2.var_names = [f"var2_{j+1}" for j in range(d2)]
adata2
/var/folders/4m/2xw3_2s503s9r616083n7w440000gn/T/ipykernel_2195/255684217.py:5: FutureWarning: X.dtype being converted to np.float32 from float64. In the next version of anndata (0.9) conversion will not be automatic. Pass dtype explicitly to avoid this warning. Pass `AnnData(X, dtype=X.dtype, ...)` to get the future behavour. adata2 = ad.AnnData(y2)
AnnData object with n_obs × n_vars = 1000 × 50
然后可以将这两个 AnnData 对象(两个“模态”)包装到单个 MuData 对象中。在这里,我们将模态一命名为A,模态二命名为B。
mdata = md.MuData({"A": adata, "B": adata2})
mdata
MuData object with n_obs × n_vars = 1000 × 150
2 modalities
A: 1000 x 100
B: 1000 x 50
MuData 对象的obs和var变量是全局的,这意味着.obs_names不同模态中具有相同名称 ( ) 的obs被认为是相同的obs。而var名称 ( .var_names) 是唯一的。在上面的对象描述中:mdata有 1000 个obs和 150 = 100+50 个var。
2.3 MuData属性¶
MuData 对象由前面描述的 AnnData 对象(如.obs或.var组成),.mod用作单个模态的访问器。
.mod模态存储在可通过MuData 对象的属性访问的集合中,其中模态名称作为键,AnnData 对象作为值。
list(mdata.mod.keys())
['A', 'B']
.mod可以通过属性或通过 MuData 对象本身作为简写来访问各个模态的名称。
print(mdata.mod["A"])
print(mdata["A"])
AnnData object with n_obs × n_vars = 1000 × 100 AnnData object with n_obs × n_vars = 1000 × 100
样本(cells)注释可通过.obs属性访问,并且默认情况下包括来自各个模式的数据帧的列的副本.obs。.var也是如此,它包含变量(features)的注释。从各个模态复制的obs列包含模态名称作为其前缀,例如 rna:n_genes。对于var列也是如此。但是,如果多种模态中存在具有相同名称的列.var(例如 n_cells),则这些列将跨模态合并,并且不添加前缀。当模态的 AnnData 对象中的这些槽发生更改时,例如添加新列或过滤掉样本(cells),必须使用.update()方法获取更改(见下文)。
可以在.obsm属性中访问样本(cells)的多维注释。例如,这可以是在所有模态上的 UMAP 坐标。
MuData 对象的形状由两个数字表示,两个数字计算为各个模态的形状之和: 一个表示obs的数量,另一个表示var的数量。
print(mdata.shape)
print(mdata.n_obs)
print(mdata.n_vars)
(1000, 150) 1000 150
默认情况下,var始终被计为单一模态,而具有相同名称的obs被计为同一obs,意味着同一个细胞具有跨多种模态的测量,var则代表了不同模态的特征值。
[adata.shape for adata in mdata.mod.values()]
[(1000, 100), (1000, 50)]
如果模态内的形状发生变化,比如adata2有相应的修改,则我们必须运行MuData.Update()将更新内容存放到MuData对象中
adata2.var_names = ["var_ad2_" + e.split("_")[1] for e in adata2.var_names]
print(f"Outdated variables names: ...,", ", ".join(mdata.var_names[-3:]))
mdata.update()
print(f"Updated variables names: ...,", ", ".join(mdata.var_names[-3:]))
Outdated variables names: ..., var2_48, var2_49, var2_50 Updated variables names: ..., var_ad2_48, var_ad2_49, var_ad2_50
这意味着,我们的MuData中存放的是对原始对象的引用,我们在更改原始对象的非结构化特征时,不用通过update一样能作用于最后的MuData
# Add some unstructured data to the original object
adata.uns["misc"] = {"adata": True}
# Access modality A via the .mod attribute
mdata.mod["A"].uns["misc"]
{'adata': True}
2.4 映射关系¶
我们在创建MuData对象时,会同时创建模态的映射关系,映射关系由bool进行存储。比如,我们模态A和模态B中所有的obs相同,那么在MuData对象中,obsm也相同
np.sum(mdata.obsm["A"]) == np.sum(mdata.obsm["B"]) == n
True
然而,对于var来说,它们是 150 长的向量。模态A有 100 个 True 值,后跟 50 个 False 值,模态B则是前面有100个False值,后面有50个True值
mdata.varm["A"]
array([ True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False])
2.5 视图¶
与 AnnData 对象的行为类似,对 MuData 对象进行切片会返回原始数据的视图。
view = mdata[:100, :1000]
print(view.is_view)
print(view["A"].is_view)
True True
对 MuData 对象进行子集化很特殊,因为它可以跨模态对它们进行切片。obs_names和var_names对每个模态执行一组和/或的切片操作,而不仅仅是针对全局多模态注释。此行为使工作流节省内存,这在处理大型数据集时尤其重要。但是,如果要修改对象,则应创建它的副本,该副本不再是视图并且不依赖于原始对象。
mdata_sub = view.copy()
mdata_sub.is_view
False
2.6 MuData对象的读写¶
与 AnnData 对象类似,MuData 对象被设计为序列化为基于 HDF5 的.h5mu文件。所有模态都以其各自的名称存储/mod在. 每个单独的模态,例如,以与存储在文件中相同的方式存储。MuData 对象可以按如下方式读写:.h5mu file/mod/A.h5ad
mdata.write("../../data/my_mudata.h5mu")
mdata_r = md.read("../../data/my_mudata.h5mu", backed=True)
mdata_r
MuData object with n_obs × n_vars = 1000 × 150 backed at '../../data/my_mudata.h5mu'
2 modalities
A: 1000 x 100
uns: 'misc'
B: 1000 x 50
MuData对象内的单模态也支持流式读取
mdata_r["A"].isbacked
True
如果备份了原始对象,则必须将文件名提供给.Copy()调用,并且生成的对象将在一个新的位置进行备份。
mdata_sub = mdata_r.copy("mdata_sub.h5mu")
print(mdata_sub.is_view)
print(mdata_sub.isbacked)
False True